A standardized platform-agnostic and future-proof format
The Standard (LASTQ) format guarantees BX:Z and VX:i SAM tags that encode the barcode and it's
validity. This format is universal and agnostic to all existing linked-read varieties and their particulars.
It is 100% compliant with comment transfer during alignment and trivial to parse once in SAM format.
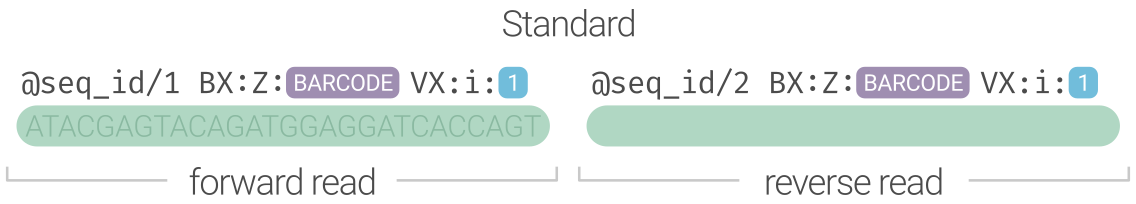
This format makes it the responsibility of the early data processing software (as early as demultiplexing) to encode the data in this format for downstream processing. Our hope is that this flexible htslib-compliant format gets widespread adoptions to reduce the fragmentation in the software ecosystem and reduce the need for file format conversions. It also creates a blueprint for a generic file encoding for any new linked-read methods that are being developed or can be developed in the future.
Specification
-
BX:Ztag to record the barcode, the format of which is irrelevant- it could be haplotagging
ACBD, stLFR1_2_3, nucleotides, whatever
- it could be haplotagging
-
VX:itag to record if the corresponding barcode is validVX tag tag value definition VX:i:00barcode is invalid VX:i:11varcode is valid -
Uses older CASAVA format (
/1or/2) to identify forward/reverse sequence- suffixed at the end of the sequence ID, without spaces
- e.g.,
@A00470:481:HNYFWDRX2:1:2101:29532:1063/1
-
Comments are SAM
TAG:TYPE:VALUEformat and tab-separated see below -
standard FASTQ naming conventions:
.fastqor.fq.fastq.gzor.fq.gzif b/gzipped.For.R1to indicate it's read 1 (forward read).Ror.R2to indicate it's read 2 (reverse read)- e.g.,
sample_1.R1.fq.gz
Despite the half-serious name LASTQ, standard-format files are FASTQ files and their naming conventions should follow FASTQ conventions exactly.
FASTQ example
stLFR style FASTQ header with a valid barcode
@A00470:481:HNYFWDRX2:1:2101:29532:1063/1 BX:Z:45_11_361 VX:i:1
haplotagging style FASTQ header with an invalid barcode
@A00470:481:HNYFWDRX2:1:2101:29532:1063/1 BX:Z:A78C00B14D96 VX:i:0
SAM/BAM example
haplotagging style SAM record with a valid barcode
A00470:481:HNYFWDRX2:1:2229:29912:29778 99 2R 1222 40 19S80M = 1500 428 AGATGTGTATAAGAGACAGAGTTATGTCATTTTAAGCGGTCAAAATGGGTGAATTTCCGATTTCAAGTAATAGGCGAACTCAAGATACCTTCTACAGAT FFFFFFFFFFFFFFFFF:FFFFF:FFFFF:FFFF:FFFFFFFFFFFFFFFFF:FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF:,FFFFFFF:FFFFF NM:i:0 MD:Z:80 MC:Z:150M AS:i:80 XS:i:80 RG:Z:sample1 MI:i:1 VX:i:1 BX:Z:A55C67B91D96
nucleotide (TELLseq/10X) style SAM record with an invalid barcode
A00470:481:HNYFWDRX2:1:2229:29912:29778 99 2R 1222 40 19S80M = 1500 428 AGATGTGTATAAGAGACAGAGTTATGTCATTTTAAGCGGTCAAAATGGGTGAATTTCCGATTTCAAGTAATAGGCGAACTCAAGATACCTTCTACAGAT FFFFFFFFFFFFFFFFF:FFFFF:FFFFF:FFFF:FFFFFFFFFFFFFFFFF:FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF:,FFFFFFF:FFFFF NM:i:0 MD:Z:80 MC:Z:150M AS:i:80 XS:i:80 RG:Z:sample1 MI:i:1 VX:i:0 BX:Z:TACANNNCACAGAG
SAM Tag Specification
Valid SAM tags take the form TAG:TYPE:VALUE, which are discussed here in further detail. The official SAM spec for tags can be found here.
TAG
All SAM tags are 2 capital alphanumeric characters, like the BX in BX:Z. HTSlib has established standard definitions for some tags (see below), which means
you should not repurpose those tags or populate them with anything other than what the SAM spec has established them to be.
TYPE
All SAM tags require a single case-sensitive character to specify
what the data type is. The most common ones you will see are Z and
i (e.g., BX:Z, VX:i). For convenience, these are the SAM spec
type definitions:
| character | data type |
|---|---|
A | character |
B | general array |
f | real number |
H | hexadecimal array |
i | integer |
Z | string |
Reserved Tags
These are the tags resereved by the SAM spec and cannot be used for anything other than what they are described for.
This also means you cannot change tag types. For example, AM:i is a standard SAM tag, which means it cannot take
any other form like AM:Z, AM:H, etc. Even if a record does not contain an AM:i tag, adding any variant of it, in
terms of data type or the meaning of the value it encodes, is very likely to break compatibility with tools that read
SAM tags.
| TAG | TYPE | Description |
|---|---|---|
| AM | i | The smallest template-independent mapping quality in the template |
| AS | i | Alignment score generated by aligner |
| BC | Z | Barcode sequence identifying the sample |
| BQ | Z | Offset to base alignment quality (BAQ) |
| BZ | Z | Phred quality of the unique molecular barcode bases in the OX tag |
| CB | Z | Cell identifier |
| CC | Z | Reference name of the next hit |
| CG | B,I | BAM only: CIGAR in BAM’s binary encoding if (and only if) it consists of >65535 operators |
| CM | i | Edit distance between the color sequence and the color reference (see also NM) |
| CO | Z | Free-text comments |
| CP | i | Leftmost coordinate of the next hit |
| CQ | Z | Color read base qualities |
| CR | Z | Cellular barcode sequence bases (uncorrected) |
| CS | Z | Color read sequence |
| CT | Z | Complete read annotation tag, used for consensus annotation dummy features |
| CY | Z | Phred quality of the cellular barcode sequence in the CR tag |
| E2 | Z | The 2nd most likely base calls |
| FI | i | The index of segment in the template |
| FS | Z | Segment suffix |
| FZ | B,S | Flow signal intensities |
| GC | ? | Reserved for backwards compatibility reasons |
| GQ | ? | Reserved for backwards compatibility reasons |
| GS | ? | Reserved for backwards compatibility reasons |
| H0 | i | Number of perfect hit |
| H1 | i | Number of 1-difference hits (see also NM) |
| H2 | i | Number of 2-difference hits |
| HI | i | Query hit index |
| IH | i | Query hit total count |
| LB | Z | Library |
| MC | Z | CIGAR string for mate/next segment |
| MD | Z | String encoding mismatched and deleted reference bases |
| MF | ? | Reserved for backwards compatibility reasons |
| MI | Z | Molecular identifier; a string that uniquely identifies the molecule from which the record was derived |
| ML | B,C | Base modification probabilities |
| MM | Z | Base modifications / methylation |
| MN | i | Length of sequence at the time MM and ML were produced |
| MQ | i | Mapping quality of the mate/next segment |
| NH | i | Number of reported alignments that contain the query in the current record |
| NM | i | Edit distance to the reference |
| OA | Z | Original alignment |
| OC | Z | Original CIGAR (deprecated; use OA instead) |
| OP | i | Original mapping position (deprecated; use OA instead) |
| OQ | Z | Original base quality |
| OX | Z | Original unique molecular barcode bases |
| PG | Z | Program |
| PQ | i | Phred likelihood of the template |
| PT | Z | Read annotations for parts of the padded read sequence |
| PU | Z | Platform unit |
| Q2 | Z | Phred quality of the mate/next segment sequence in the R2 tag |
| QT | Z | Phred quality of the sample barcode sequence in the BC tag |
| QX | Z | Quality score of the unique molecular identifier in the RX tag |
| R2 | Z | Sequence of the mate/next segment in the template |
| RG | Z | Read group |
| RT | ? | Reserved for backwards compatibility reasons |
| RX | Z | Sequence bases of the (possibly corrected) unique molecular identifier |
| S2 | ? | Reserved for backwards compatibility reasons |
| SA | Z | Other canonical alignments in a chimeric alignment |
| SM | i | Template-independent mapping quality |
| SQ | ? | Reserved for backwards compatibility reasons |
| TC | i | The number of segments in the template |
| TS | A | Transcript strand |
| U2 | Z | Phred probability of the 2nd call being wrong conditional on the best being wrong |
| UQ | i | Phred likelihood of the segment, conditional on the mapping being correct |