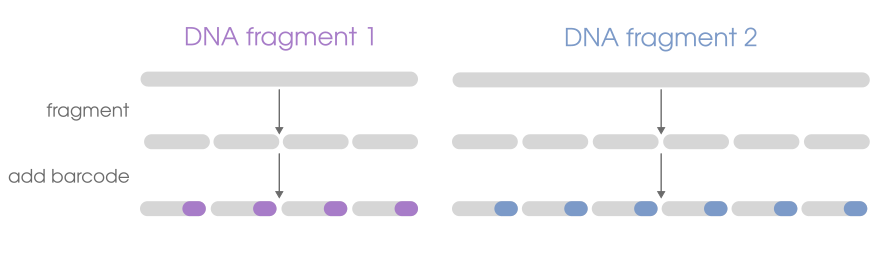
Linked reads are short-read (e.g. Illumina) data. What makes them different from other kinds of sequencing data is that they contain an added DNA segment ("barcode") that lets you associate sequences as having originated from a single DNA molecule. That means 4 sequences that all contain the same linked-read barcode can be inferred as having originated from the same original DNA molecule. Different barcode = different molecule of origin. If the sequences with the same barcode would map near the same genomic region during sequence alignment, we would know that each of those reads originated from a single DNA fragment from a single homologous chromosome from a single cell, which is effectively built-in phase information.
What do they look like?
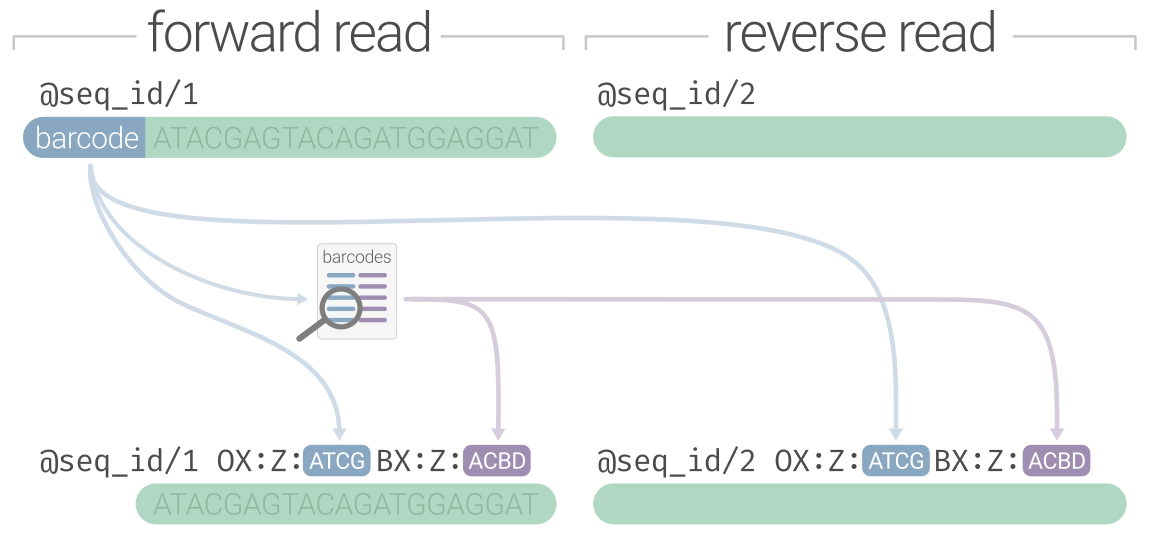
Linked-read sequence data appears as you might expect it, encoded in a FASTQ file. The first processing step of
linked-read data is demultiplexing to split the raw Illumina-generated batch FASTQ file into samples (if multisample)
and identify/validate the linked-read barcode on every sequence. For 10X data, the barcode would stay inline with
the sequence (to make it LongRanger compatible), but for other varieties (haplotagging, stLFR, etc.) you would also
remove the barcode from the sequence and preserve it somewhere in the read header. The demultiplexing process
is generally similar between non-10X linked-read technologies: a nucleotide barcode sequence gets identified and moved from
the sequence line to the read header with some kind of platform-specific notation. The diagram below preserves the nucleotide
barcode under the OX:Z tag and recodes it under BX:Z using the haplotagging "ACBD" segment format, however it would
also be valid to just keep the nucleotide barcode under BX:Z. Linked-read software is variable in its flexibility towards barcode
formatting.
Linked-read varieties
There are a handful of linked-read sample preparation methods, but that's largely an implementation detail. All of those methods are laboratory procedures to take genomic DNA and do the necessary modifications to fragment long DNA molecules, tag the resulting fragments with the same DNA barcode, then add the necessary Illumina adapters. It's not unlike the different RAD flavors (e.g. EZrad, ddRAD, 2B-rad)-- they all give you RAD data in the end, but vary in how you get there in terms of cost and bench time.
Always Usable Data
Unlike some other sample preparation methods (cough cough mate-pair), linked-read data is whole genome sequencing (WGS). What that means is that whether you use the linked-read information or not, the data will always be standard and viable WGS compatible with whatever you would use WGS for. It's WGS, but with a little extra info that goes a long way.