Intro to linked-read data
Harpy was originally tailor-made for haplotagging linked read data, but now it works for most linked-read technologies along with non-linked read data. BRL/LRTK/LongRanger are similar pieces of software for Tellseq, stLFR, and 10X linked-read data. But, what if you don't use linked reads (yet) and want to understand what it actually is? This post walks you through what linked-read data is and some of the concepts that are unique to it that makes it different than your typical short-read data.
- a cup of tea, coffee, or water
- a can-do attitude
Linked reads
Let's start with the most obvious, what are linked reads, which are sometimes called "synthetic long reads"? They are short read
(e.g. Illumina) data. What makes them different is that they contain an added DNA segment ("barcode") that lets us associate
sequences as having originated from a single DNA molecule. That means if we have 4 sequences that all contain the same added barcode,
then we infer that they must have originated from the same original DNA molecule. Different barcode = different molecule of origin.
If the sequences would map to the same genomic region during sequence alignment, we would know that those sequences with the same
barcode originated from a single DNA fragment from a single homologous chromosome from a single cell. That's right, built-in phase information.

Linked-read data is sequence data as you would expect it, encoded in a FASTQ file. The first processing step of
linked-read data is demultiplexing to split the raw Illumina-generated batch FASTQ file into samples (if multisample)
and identify/validate the linked-read barcode on every sequence. For 10X data, the barcode would stay inline with
the sequence (to make it LongRanger compatible), but for other varieties (haplotagging, stLFR, etc.) you would also
remove the barcode from the sequence and preserve it somewhere in the read header. The demultiplexing process
is generally similar between non-10X linked-read technologies: a nucleotide barcode sequence gets identified and moved from
the sequence line to the read header with some kind of platform-specific notation. The diagram below preserves the nucleotide
barcode under the OX:Z tag and recodes it under BX:Z using the haplotagging "ACBD" segment format, however it would
also be valid to just keep the nucleotide barcode under BX:Z. Linked-read software is variable in its flexibility towards barcode
formatting. 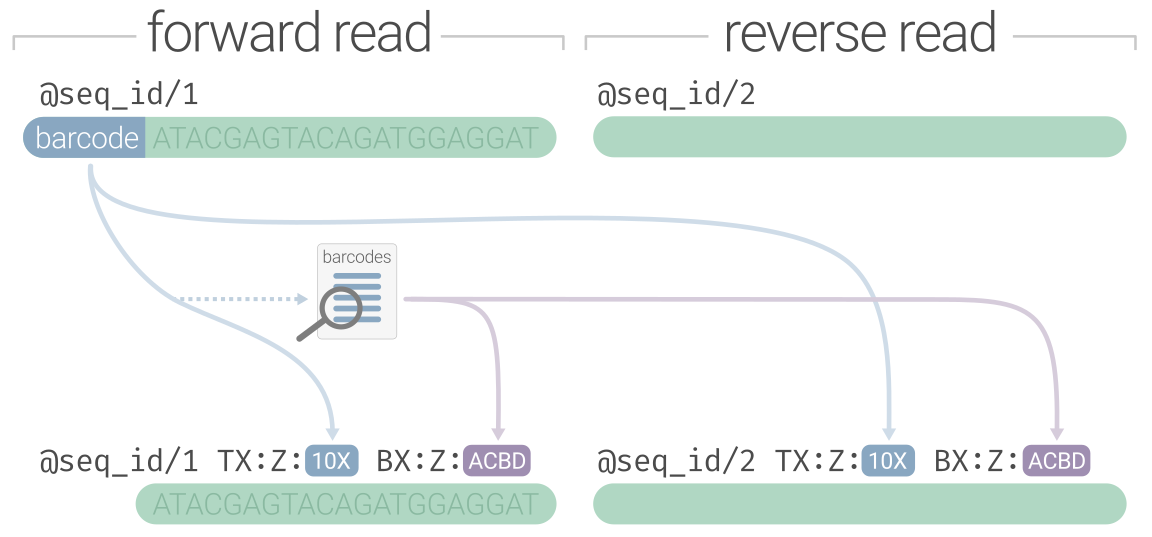
There are a handful of linked-read sample preparation methods, but that's largely an implementation detail. All of those methods are laboratory procedures to take genomic DNA and do the necessary modifications to fragment long DNA molecules, tag the resulting fragments with the same DNA barcode, then add the necessary Illumina adapters. It's not unlike the different RAD flavors (e.g. EZrad, ddRAD, 2B-rad)-- they all give you RAD data in the end, but vary in how you get there in terms of cost and bench time. We obviously subscribe to haplotagging 😁.
It's worth describing the obvious differences of the raw (FASTQ) data. Knowing these details might help you make sense of compatibilties/incompatibilities for software, or how you can convert between styles. For more information, read this resource. We strongly advocate for using Standard format.
Unlike some other fancy well-touted sample preparation methods (cough cough mate-pair), linked-read data is whole genome sequencing (WGS). What that means is that whether you use the linked-read information or not, the data will always be standard and viable WGS compatible with whatever you would use WGS for. It's WGS, but with a little extra info that goes a long way.
There are approaches to deconvolve linked read data (or "deconvolute", if you're indifferent to the burden of an extra syllable).
In this context, deconvolution is finding out which sequences are sharing barcodes by chance rather than because they originated
from the same DNA molecule. The likelihood of it happening is usually low, but it's not impossible. When the algorithm determines
that barcodes are being shared by unrelated molecules, the barcode typically gets suffixed with a hyphenated number (e.g.
BX:Z:ATACG becomes BX:Z:ATACG-1). As of this writing, there are only a few pieces of software that can deconvolve linked-read data and
do so with varying degrees of success and computational resource requirements. Similarly, linked-read software is variable in its
flexibility towards accepting the deconvolved-barcode format.
Linked-read library performance
Because linked-read data has an extra dimension, there are some additional metrics that are useful to look at to evaluate how "good" the linked-read library turned out. As a baseline for comparison, think of RAD or WGS data and the kinds of metrics you might look at for library performance:
- coverage depth
- coverage breadth
- PCR/optical duplicates
- coverage depth per sample
Linked-read library performance also looks at that, but there's a few extra parameters that help us assess performance:
Molecule Coverage
Since linked reads are tagged per DNA molecule, we are interested in understanding coverage on a per-molecule basis. By "molecule", we are referring to the original DNA molecule from which barcoded sequences originated from.
It's quite rare that all fragments of a single DNA molecule will get sequenced, so we are interested in getting an average number of reads (sequences) per original DNA molecule. This is done by getting counts of all the reads with the same barcode, as unique barcodes correspond with unique DNA molecules. It's difficult to say exactly what a good number of reads per molecule would be, but >2 would be a good starting point (1 would be a singleton, see below). Having 3-6 would be decent, depending on your project goals.
Because only a few fragments from a DNA molecule end up getting sequenced, it would be good to understand how much of a molecule is
represented in seqences (breadth). It's impossible to get an accurate calculation for this metric because we have no way of knowing
the size of the original DNA molecule, but what we can do is, after alignment, find the distance between the two furthest reads
sharing a barcode and calculate how many bases between them have been sequenced. It's not perfect, but it's something. A low number
would indicate large gaps between linked sequences (e.g. few sequences and/or a very large molecule), whereas a high number would
indicate small gaps between linked sequences (e.g. many sequences and/or a very small molecule).

Singletons
Because linked reads need to be, well, linked, we need to know exactly how many of the sequences actually share barcodes. A barcode
that only appears in one paired or unpaired read is a singleton, meaning it isn't actually linked to any other sequence. Despite
having a linked-read barcode, the absence of other reads with the same barcode means the barcode information for that read (or read
pair) is mostly useless, aka it's just a plain-regular short read and can be used as such. Having <60% would be considered decent,
but you would want as few singletons as possible in a linked-read dataset. For perspective, if 90% of your reads were singletons, you
can think of that as "90% of your data aren't linked reads". The opposite of a singleton is when a barcode is shared by more than one
paired or unpaired read, aka a linked read.

Linked Coverage
Because of the "linked" component of linked-read data, we have an additional kind of sequence coverage
to consider, which is linked coverage. Since linked-read barcodes preserve long-distance information, we can do an alternative kind of
coverage calculation where you pretend that all the sequences with a shared barcode are one big gapless sequence. If you use your
imagination that way, the name "synthetic long reads" begins to make more sense. Mathematically, the linked coverage (breadth and
depth) should always be higher than the coverage from just the alignments themselves, as gaps between linked reads are counted as
well. If you see that your linked depth is unusually high (e.g. the aligmment depth is 20 and linked depth is >1000), then it's
very likely your have barcode clashing that has not been deconvoluted. In other words, reads from different DNA molecules that are
sharing a barcode are mapped very far from each other and inflating the depth values for everything in between.

Barcode thresholds
By the nature of linked read technologies, there will (almost always) be more DNA fragments than unique barcodes for them. As a result,
it's common for barcodes to reappear in sequences. Rather than incorrectly assume that all sequences/alignments with the same barcode
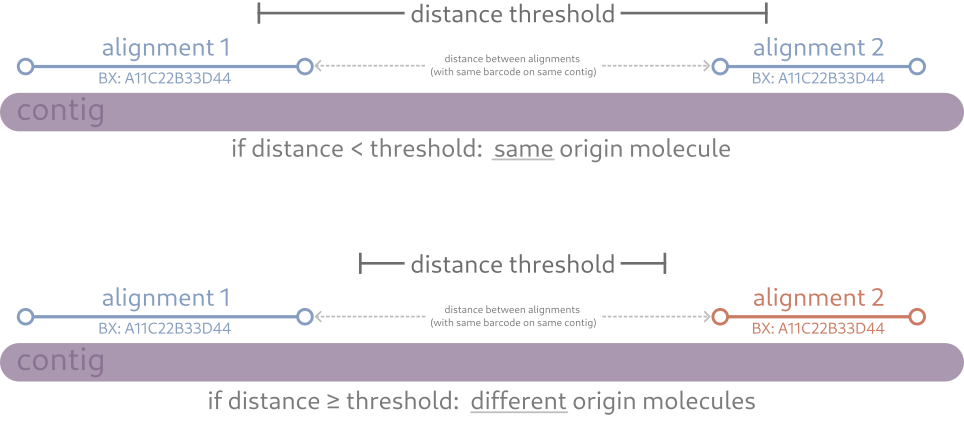
originated from the same orignal DNA molecule, linked-read aware programs include a threshold parameter to determine a "cutoff" distance
between alignments with the same barcode. This parameter can be interpreted as "if a barcode appears more than X base pairs away from the
same barcode (on the same contig), then we'll consider them as originating from different molecules." If this threshold is lower, then
you are being more strict and indicating that alignments sharing barcodes must be closer together to be considered originating from the same
DNA molecule. Conversely, a higher threshold indicates you are being more lax and indicating barcodes can be further away from each other
and still be considered originating from the same DNA molecule. A threshold of 50kb-150kb is considered a decent balance, but you should choose
larger/smaller values if you have evidence to support them.
Potential SV detection obstruction
This kind of deconvolving method relies on alignment distances, which may worsen performance in finding structural variants larger than this threshold. For example, two reads originating from the same DNA molecule may have aligned quite far from each other due to mapping along the breakpoint of a very large inversion. If the inversion spans 3Mb, a barcode threshold of 100kb will likely incorrectly deconvolve the two reads and assume they shared a barcode by chance, which would hurt SV detection performance.