align bwa
- at least 4 cores/threads available
- a genome assembly in FASTA format: .fasta .fa .fasta.gz .fa.gz case insensitive
- paired-end fastq sequence files
❤️
gzipped recommended
- sample name: a-z 0-9 . _ - case insensitive
- forward: _F .F .1 _1 _R1_001 .R1_001 _R1 .R1
- reverse: _R .R .2 _2 _R2_001 .R2_001 _R2 .R2
- fastq extension: .fq .fastq case insensitive
Once sequences have been trimmed and passed through other QC filters, they will need to be aligned to a reference genome. This module within Harpy expects filtered reads as input, such as those derived using harpy qc . You can map reads onto a genome assembly with Harpy using the align bwa module:
harpy align bwa OPTIONS... REFERENCE INPUTS...harpy align bwa genome.fasta Sequences/ Running Options
In addition to the common runtime options , the align bwa module is configured using these command-line arguments:
Output format
Regardless of the input linked-read format, the align workflows will standardize the output alignment records
such that the barcode is contained in the BX:Z tag and barcode validation is in the VX:i tag.
Molecule distance
The --molecule-distance option is used during the alignment workflow
to deconvolute alignments with the same barcode that might not have originated
from the same DNA molecule based on the distance threshold
you specify. This happens during the linked-read stats step to internally split molecules based on this value, but
it doesn't modify the barcodes in the output. Set this value to 0 to skip distance-based deconvolution during the
this reporting step. Ignored if using --skip-reports.
Quality filtering
The --min-quality argument filters out alignments below a given MQ threshold. The default, 30, keeps alignments
that are at least 99.9% likely correctly mapped. Set this value to 1 if you only want alignments removed with
MQ = 0 (0% likely correct). You may also set it to 0 to keep all alignments for diagnostic purposes.
The plot below shows the relationship between MQ score and the likelihood the alignment is correct and will serve to help you decide
on a value you may want to use. It is common to remove alignments with MQ <30 (<99.9% chance correct) or MQ <40 (<99.99% chance correct).
Every alignment in a BAM file has an associated mapping quality score (MQ) that informs you of the likelihood that the alignment is accurate. This score can range from 0-40, where higher numbers mean the alignment is more likely correct. The math governing the MQ score actually calculates the percent chance the alignment is incorrect:
\%\ chance\ incorrect = 10^\frac{-MQ}{10} \times 100\\
\text{where }0\le MQ\le 40You can simply subtract it from 100 to determine the percent chance the alignment is correct:
\%\ chance\ correct = 100 - \%\ chance\ incorrect\\
\text{or} \\
\%\ chance\ correct = (1 - 10^\frac{-MQ}{10}) \times 100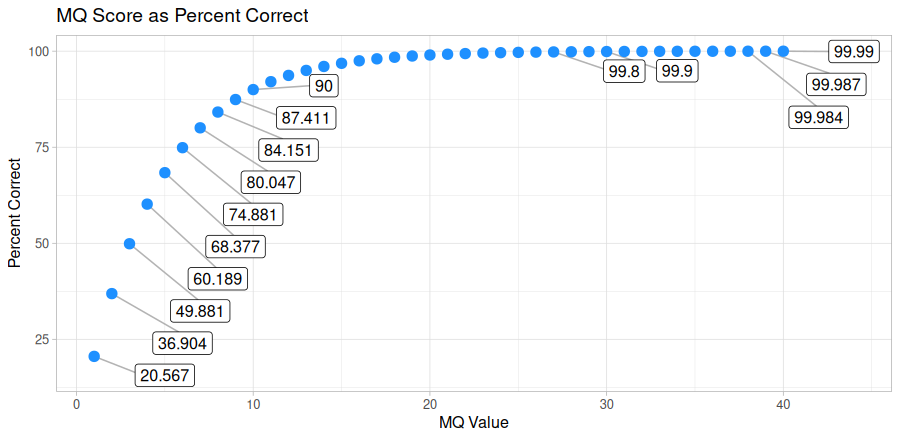
Marking PCR duplicates
Harpy uses samtools markdup to mark putative PCR duplicates by using both the BX tag
as a UMI (unique molecule identified) for more accurate duplicate detection. The read name
is also parsed to determine if the sequencing platform was HiSeq/NovaSeq to distinguish between
PCR and optical duplicates. Duplicate marking also uses the -S option to mark supplementary (chimeric)
alignments as duplicates if the primary alignment was marked as a duplicate. Duplicates get marked but are not removed.
BWA workflow
- ignores (but retains) barcode information
- fast
The BWA MEM workflow maps all reads against the reference genome. Duplicates are marked using samtools markdup.
The BX:Z tags in the read headers are still added to the alignment headers, even though barcodes
are not used to inform mapping. The -m threshold is used for alignment molecule assignment.
graph LR
A([index genome]):::clean --> B([align to genome]):::clean
B-->C([sort alignments]):::clean
C-->XX([standardize barcodes]):::clean
XX-->D([mark duplicates]):::clean
D-->E([assign molecules]):::clean
E-->F([alignment metrics]):::clean
D-->G([barcode stats]):::clean
G-->F
subgraph aln [Inputs]
Z[FASTQ files]:::clean---genome:::clean
end
aln-->B & A
subgraph markdp [mark duplicates via `samtools`]
direction LR
collate:::clean-->fixmate:::clean
fixmate-->sort:::clean
sort-->markdup:::clean
end
style markdp fill:#f0f0f0,stroke:#e8e8e8,stroke-width:2px,rx:10px,ry:10px
style aln fill:#f0f0f0,stroke:#e8e8e8,stroke-width:2px,rx:10px,ry:10px
classDef clean fill:#f5f6f9,stroke:#b7c9ef,stroke-width:2px
The default output directory is Align/bwa with the folder structure below. Sample1 is a generic sample name for demonstration purposes.
The resulting folder also includes a workflow directory (not shown) with workflow-relevant runtime files and information.
Align/bwa
├── Sample1.bam
├── Sample1.bam.bai
├── logs
│ ├── sample1.bwa.log
│ ├── sample1.markdup.log
│ │── sample1.sort.log
└── reports
├── barcodes.summary.html
├── bwa.stats.html
├── Sample1.html
└── data
├── bxstats
│ └── Sample1.bxstats.gz
└── coverage
└── Sample1.cov.gzBy default, Harpy runs bwa with these parameters (excluding inputs and outputs):
bwa-mem2 mem -v 2 -T 10 -m 10 -C -R "@RG\tID:samplename\tSM:samplename"Below is a list of all bwa-mem2 mem command line arguments, excluding those Harpy already uses or those made redundant by Harpy's implementation of BWA.
These are taken directly from the BWA documentation.
Algorithm options:
-k INT minimum seed length [19]
-w INT band width for banded alignment [100]
-d INT off-diagonal X-dropoff [100]
-r FLOAT look for internal seeds inside a seed longer than {-k} * FLOAT [1.5]
-y INT seed occurrence for the 3rd round seeding [20]
-c INT skip seeds with more than INT occurrences [500]
-D FLOAT drop chains shorter than FLOAT fraction of the longest overlapping chain [0.50]
-W INT discard a chain if seeded bases shorter than INT [0]
-S skip mate rescue
-P skip pairing; mate rescue performed unless -S also in use
Scoring options:
-A INT score for a sequence match, which scales options -TdBOELU unless overridden [1]
-B INT penalty for a mismatch [4]
-O INT[,INT] gap open penalties for deletions and insertions [6,6]
-E INT[,INT] gap extension penalty; a gap of size k cost '{-O} + {-E}*k' [1,1]
-L INT[,INT] penalty for 5'- and 3'-end clipping [5,5]
-U INT penalty for an unpaired read pair [17]
Input/output options:
-p smart pairing (ignoring in2.fq)
-H STR/FILE insert STR to header if it starts with @; or insert lines in FILE [null]
-j treat ALT contigs as part of the primary assembly (i.e. ignore <idxbase>.alt file)
-5 for split alignment, take the alignment with the smallest coordinate as primary
-q don't modify mapQ of supplementary alignments
-K INT process INT input bases in each batch regardless of nThreads (for reproducibility) []
-h INT[,INT] if there are <INT hits with score >80% of the max score, output all in XA [5,200]
-a output all alignments for SE or unpaired PE
-V output the reference FASTA header in the XR tag
-Y use soft clipping for supplementary alignments
-M mark shorter split hits as secondary
-I FLOAT[,FLOAT[,INT[,INT]]]
specify the mean, standard deviation (10% of the mean if absent), max
(4 sigma from the mean if absent) and min of the insert size distribution.
FR orientation only. [inferred]