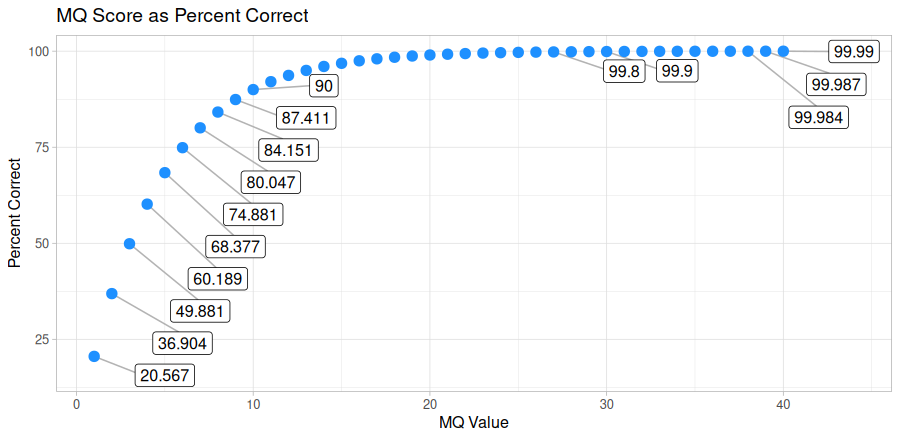
You may want to phase your genotypes into haplotypes, as haplotypes tend to be more informative than unphased genotypes (higher polymorphism, captures relationship between genotypes). Phasing genotypes into haplotypes requires alignment files, such as those produced by and a variant call file, such as one produced by or . Phasing only works on SNP/indel data, and will not work for structural variants produced by or , preferably filtered in some capacity. You can phase genotypes into haplotypes with Harpy using the module: