Background
Structure is a brilliant intellectual work. Structure is also not user-friendly. In fact, one might go so far as to say it’s sometimes user-hostile.
User hostility
- extremely unforgiving input file parsing
- manual is not terribly user friendly
- program is not user friendly
- GUI version isn’t great for long runs (i.e. modern data)
- two different parameter files
- there are a million parameters
- default plots suck and aren’t dichromat friendly
- runs on a single thread/cpu and takes a while depending on your data
So, using Structure is largely about navigating these pitfalls. First, we’re going to ignore the GUI altogether and run Structure via command line, which comes with the added bonus of being able to run multiple instances of Structure at once for different parameters (like K). This needs to be done in either macOS or Linux. Since it’s a C/Java program, I’m sure it can on Windows too, but I never bothered trying. Using cmd or powershell in Windows is a personal hell.
Requirements
- Structure Downloaded from here (a conda installation probably works too)
- Java installed on the system if using the GUI (we aren’t)
- Patience
Input Files
Structure has a very specific file format and it’s awful. The first row is the locus names (optional), and the subsequent rows are the sample names (optional) and their genotypes, space delimited. You can also have extra rows after the locus header, and you’ll need to specify how many extra rows in the “params” files later if you do. After the header (if it’s there), each sample gets ploidy number of rows (or not), one for each allele per locus. You can force it to be one sample per row, but I assure you that’s a nightmare waiting to happen for non-haploids. You see why this is frustrating? An example of a real-world Structure input file can be found here. Snippets of it will be used for explanations below.
Example input file (diploid)
snp_1 snp_2 snp_3 <--- header of loci
ind_001 002 -9 001 <--- sample ind_001, first allele per locus
ind_001 001 -9 002 <--- sample ind_001, second allele per locus
ind_002 001 001 001 <--- sample ind_002, first allele per locus
ind_002 002 002 001 <--- sample ind_002, second allele per locus
The <--- some words are for teaching purposes, don’t actually include that in your input file or Structure will yell at you. The file continues like that until the very end. Missing genotypes are traditionally coded as -9 (no idea why) , and you can change that in the mainparams with the MISSING parameter. Pro tip: just keep missing as -9.
Extra Columns
Unlike extra rows (which probably have some use somewhere), extra columns are very important for the input files. Extra columns go in between the sample name and the first genotype and have to be noted in the params files (explained below). Here is an example of a snippet of a real-world structure file with extra columns:
snp_1 snp2 snp_3
cca_001 1 1 1 002 001 001
cca_001 1 1 1 001 001 002
cca_002 1 1 1 001 001 001
cca_002 1 1 1 002 002 001
gaa_001 1 1 2 002 001 001
gaa_001 1 1 2 001 001 001
gaa_002 1 1 2 001 001 001
gaa_002 1 1 2 001 001 001
key_001 2 0 3 001 002 001
key_001 2 0 3 001 002 001
key_002 2 0 3 001 001 001
key_002 2 0 3 001 001 002
meg_002 2 1 4 001 001 001
meg_002 2 1 4 001 002 001
meg_003 2 1 4 001 001 001
meg_003 2 1 4 002 001 002
seg_001 2 1 5 001 -9 -9
seg_001 2 1 5 001 -9 -9
seg_003 2 1 5 001 001 001
seg_003 2 1 5 001 001 001
These three extra columns correspond to these parameters (descriptions stolen from official docs):
First extra column: POPDATA
An integer designating a user-defined population from which the individual was obtained (for instance these might designate the geographic sampling locations of individuals). In the default models, this information is not used by the clustering algorithm, but can be used to help organize the output (for example, plotting individuals from the same pre-defined population next to each other).
The human version:
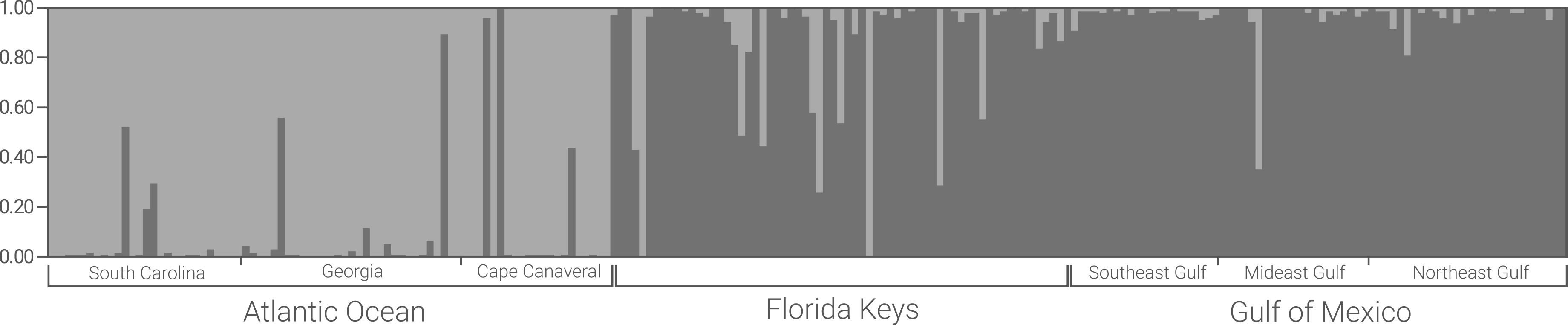
It’s some sort of population pre-designation which Structure won’t use for analysis unless you tell it to. In the example file above, 1 indicates “Atlantic Ocean” and 2 indicates “Gulf of Mexico”.
Second extra column: POPFLAG
A Boolean flag (0 or 1) which indicates whether to use the PopData when using learning samples.
The human version:
If doing assignment testing for an unknown group, this is super important. In the file above, I wanted to see what population (Atlantic or Gulf) the Florida Keys samples (key_) assigned to, so I’m omitting them from “teaching” Structure what Atlantic or Gulf populations “look” like by setting that second extra column to 0. You can see that the cca_ and gaa_ samples (Atlantic) have a 1, as do the meg_ and seg_ samples (Gulf), meaning they will be included in establishing my population signals before assignment. This now means the key_ samples will later be assigned to these populations.
Third extra column: LOCDATA
An integer designating a user-defined sampling location (or other characteristic, such as a shared phenotype) for each individual. This information is used to assist the clustering when the LOCPRIOR model is turned on. If you simply wish to use the PopData for the LOCPRIOR model, then you can omit the LocData column and set LOCISPOP=1 (this tells the program to use PopData to set the locations).
The human version:
This is extra information to let you make stuff more hierarchical, of sorts. Although I have my POPDATA designations of Atlantic/Gulf, I use the LOCDATA column to break the samples down into finer sampling localities. So, cca_ (Cape Canaveral), is one location, meg_ (Mideast Gulf) is another, key_ (Florida Keys) is another, etc.
Creating the input file
Options include PGDSpider2, R::radiator, and probably some other ones. You don’t want to create this file by hand. If you already have a Genepop formatted file on hand, I made a script that converts it into Structure format via radiator here. You can also use PopGen.jl to read in from genepop/vcf/plink/csv and write to structure of faststructure formats.
Parameter files
You can run Structure and manually specify every parameter as arguments to the command line call, but absolutely don’t do that. Instead, use mainparams and extraparams files.
mainparams
Here is what your mainparams file should look like (yes, including the #define part):
#define OUTFILE some_output_name
#define INFILE some_input_file.str
#define NUMINDS some_integer
#define NUMLOCI some_integer
#define LABEL 1
#define POPDATA 1
#define POPFLAG 1
#define LOCDATA 1
#define PHENOTYPE 0
#define MARKERNAMES 1
#define MAPDISTANCES 0
#define ONEROWPERIND 0
#define PHASEINFO 0
#define PHASED 0
#define RECESSIVEALLELES 0
#define EXTRACOLS 0
#define MISSING -9
#define PLOIDY 2
#define MAXPOPS 2
#define BURNIN 1000000
#define NUMREPS 1000000
Let’s go through them:
| mainparam | what it does |
|---|---|
OUTFILE | is the name you want the output file to have (no spaces) . Don’t use quotes. |
INFILE | the name of your input file. Also no spaces, also don’t use quotes. |
NUMINDS | the number of samples in your input file. Structure refuses to intuit this number, so you have to specify it. A lot of dumb errors happen because of this. |
NUMLOCI | the number of loci in your input file. Structure also refuses to intuit this number, so you have to specify it. A lot of dumb errors happen because of this. |
LABEL | yes (1) or no (0) of whether your input file contains sample names. The example input files above would use LABEL 1 |
POPDATA, POPFLAG, and LOCDATA | are yes (1) or no (0) and we described in the previous section. We would set each of these to 1 for the example input file above |
PHENOTYPE | if phenotype info is included. Honestly never used this and don’t know how to. |
MARKERNAMES | yes (1) or no (0) of whether your input file contains a header of locus names. The example input files above would use MARKERNAMES 1 |
MAPDISTANCES | whether you have an extra row of map distances between neighboring loci. |
ONEROWPERIND | yes (1) or no (0) of whether you have a single row per sample. The example input files above would use ONEROWPERIND 0. It’s iterally the worst format ever, don’t do this. |
PHASEINFO | yes (1) or no (0) of whether you have an extra row of haplotype phase information after each sample row. This is exclusive to the other linkage-aware stuff Structure can do |
PHASED | yes (1) or no (0) of whether your haplotypes are correctly phased, whatever that means. This is exclusive to the other linkage-aware stuff Structure can do and used in tandem with PHASEINFO |
RECESSIVEALLELES | indicates the next row of the data file contains a list of L integers indicating which alleles are recessive at each locus. Setting this to 1 implies that the dominant marker model is in use. Never used this. Would be 0 for the examples above |
EXTRACOLS | the number of extra columns unrelated to Structure that the software should ignore. I would avoid adding anything you don’t need to Structure input files. |
MISSING | the value missing genotypes/alleles are coded as |
PLOIDY | the ploidy of your data. Structure needs this to read the rows correctly. It might have a math component too. Who knows. |
MAXPOPS | the number of groups Structure will try to cluster your data into. This parameter name is kind of misleading because it’s a fixed number, not iteratively trying 1:MAXPOPS. See the section below about adjusting MAXPOPS |
BURNIN | the number of iterations to use at the burn in for the Bayesian model. Ideally you’d want the model to already converge by the time it finishes the burn in, so you’d want this to be a reasonably high number. 1 million in the example above was probably overkill for the data it was used on (212 samples X 2204 snp loci) |
NUMREPS | the number of iterations to run the model after the burn in completes. You also want this to be a reasonably high number. 1 million in the example above was overkill for the data it was used on, 500k or 750k was probably enough. Structure took 22-24hrs to complete with those BURNIN and NUMREPS parameters on the 212x2204 data. |
There are a few more advanced “main” parameters, see the documentation for more information.
extraparams
If you thought the mainparams were exhaustive, well, the extraparams are even more so. This file contains parameters that govern the kind of models Structure runs and how it runs them. Here is what the extraparams file looks like:
#define NOADMIX 1
#define LINKAGE 0
#define USEPOPINFO 1
#define GENSBACK 2
#define LOCPRIOR 0
#define LOCPRIORINIT 1.0 //(d) initial value for r, the location prior
#define MAXLOCPRIOR 100.0 //(d) max allowed value for r
#define INFERALPHA 1
#define ALPHA 1.0
#define POPALPHAS 0
#define UNIFPRIORALPHA 1
#define ALPHAMAX 10.0
#define ALPHAPROPSD 0.025
#define FREQSCORR 1
#define ONEFST 0
#define FPRIORMEAN 0.01
#define FPRIORSD 0.05
#define INFERLAMBDA 0
#define LAMBDA 1.0
#define COMPUTEPROB 1
#define PFROMPOPFLAGONLY 0
#define ANCESTDIST 0
#define STARTATPOPINFO 0
#define METROFREQ 10
#define UPDATEFREQ 5000
These parameters are super specific and we’re only going to cover a few of them, so I would recommend looking at the the documentation to make sense of it all.
| extraparam | what it does |
|---|---|
NOADMIX | yes (1) or no (0) of whether to use the admixture model, i.e. are we allowing the model to consider admixed individuals (1) or can we say every individual is purely from a single population (0) |
LINKAGE | yes (1) or no (0) of whether to incorporate the linkage information mentioned above |
USEPOPINFO | yes (1) or no (0) of using the POPINFO model (corresponds to the POPFLAG above). This model uses sampling locations to test for migrants or hybrids |
GENSBACK | an integer of how many generations back to make assignment inferences for. In the example extraparams, this is set to 2, meaning we want ancestry inferred for parents and grandparents too. I’ve had minimal luck getting meaningful results from this. Use 0 to ignore this altogether. |
LOCPRIOR | a yes (1) or no (0) of using the LOCPRIOR model. Use sampling locations as prior information to assist the clustering–for use with data sets where the signal of structure is relatively weak. Location information can sometimes be the thing that gives Structure enough power to identify clusters. |
UPDATEFREQ | an integer that prints some information in fixed intervals on the screen as the program runs. Think of it like a progress bar. |
The Structure docs say it’s generally ok to try running the program without tweaking those specific model parameters like the alphas, lambda, and priors.
Optimizing K
Since Structure can’t tell you what the best K (MAXPOPS) is, you need to find that out yourself (yay). To do that, you need to run Structure for a range of MAXPOPS values and compare their likelihoods. According to the scrolls, the run with the highest likelihood is the optimal K, but take that with a grain of salt because you can’t run Structure for K=1 (panmixia might be what’s happening), and minimal differences in likelihoods obscure K optimization. See “An advanced case” below for a quick tip.
Running the program
The session
If running it from the command line, which you should, it’s generally pretty easy. For starters, since the run/runs may take a while, you don’t want to be married to a terminal session, so I recommend these options in descending order of preference:
- using
screenortmuxto detach and reattach sessions as needed nohup .... &to have it run silently without interruptions in the background- or
ctrl + zfollowed bybgthendisown -h
Actually running it
path/to/structure -m mainparams.file -e extraparams.file
Structure lets you add command line arguments to override some of the params in the files. I generally don’t recommend doing this; keeping distinct copies of param files with organized naming schemes will definitely help future-proof your work. An exception to the rule is:
-Kchange theMAXPOPSparameter- e.g.
structure -K 5 -m mainparams.file -e extraparams.file
- e.g.
-Dset the randomization seed for the run- this is useful if you want exact reproducible results
- e.g.
structure -D 6969 -m mainparams.file -e extraparams.file
An advanced case
Structure itself runs on a single thread, but you can run multiple instances of it at a time with tweaked parameters. This is very useful when you’re trying to optimize K (MAXPOPS). One quick way to do that as a one-liner just uses GNU parallel:
seq 2 9 | parallel -j 8 path/to/structure -K {} -m mainparams.file -e extraparams.file
seq n1 n2creates a sequence of numbers fromn1ton2, which is 2:9 in this example- this gets piped into
parallel, where{}is the placeholder to be replaced by this sequence
- this gets piped into
-jsets the maximum number of jobs/threads, which is 8 in this example-Kis thatMAXPOPSparameter we can overwrite
This simple syntax will have parallel run -K 2 through -K 9 with up to 8 of them at a time (which is all of them!) 💃
Visualizing the results
Options include:
- Using the Structure GUI to load in and visualize your results for basic data exploration.
- life hack: save as a .pdf and open in Illustrator/Inkscape to manually edit colors
- Apparently this website is a thing
- This script seems promising too and is probably what I’d lean towards outside of writing my own
- Of course I wrote my own script! Here is where it lives
Troubleshooting
Most of the woes with Structure are just getting the input file and params to agree with each other.
If using a Windows machine in any part of preparing your data for Structure:
Play it safe and run the file through the GNU command line tool dos2unix. Sometimes errors happen just because of the Windows CRLF line endings. When in doubt, make sure your files(s) use the Unix “LF” endings (default for macOS, Linux, BSD) and use UT-8 encoding (default for most things).